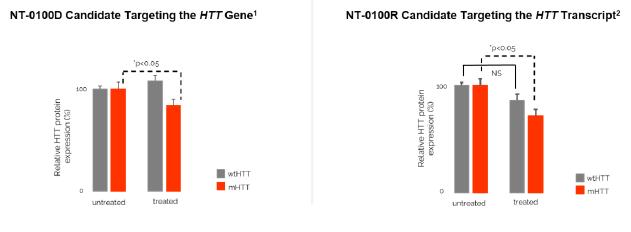
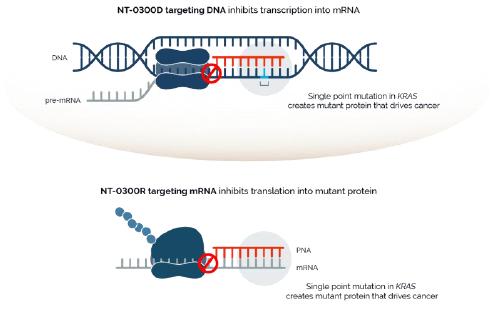
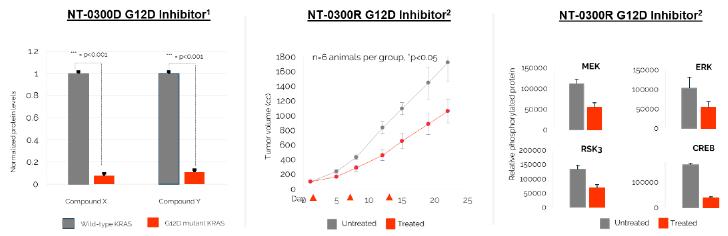
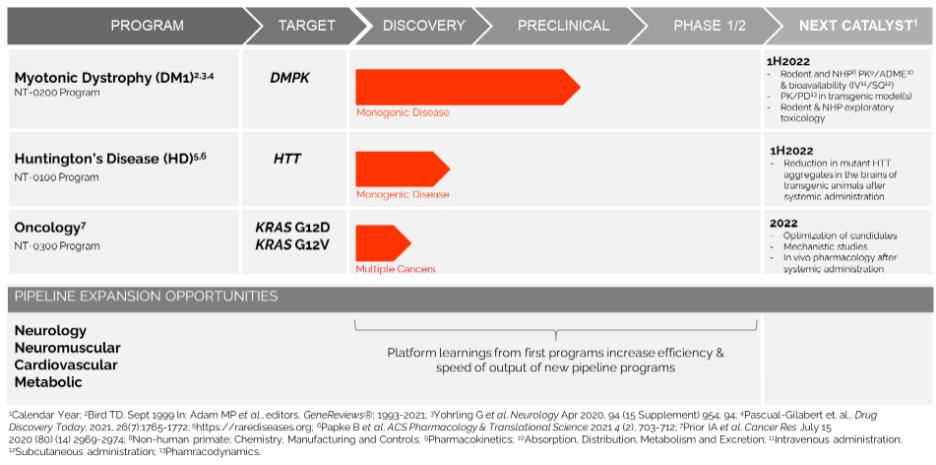
In addition, pursuant to the Offer of Employment, Dr. Stephan’s confidential information, invention assignment and arbitration agreement with Legacy NeuBase shall continue to apply and was assumed by us.
On May 15, 2020, the Company awarded a discretionary cash bonus to Dr. Stephan in the amount of $157,500 for services rendered during the fiscal year ended September 30, 2019, which were not committed during such fiscal year and were approved by the Compensation Committee of the Board during May 2020.
Sam Backenroth
On January 6, 2015, Ohr amended its employment agreement with Sam Backenroth, Chief Financial Officer and Vice President, Business Development, to extend the term to February 28, 2016, and to provide for automatic one year extensions thereafter absent notice of termination. The employment agreement provided for an annual base salary of $200,000 for Mr. Backenroth.
Mr. Backenroth was entitled to (1) severance pay and benefits if his employment is terminated, whether at the end of the term of his employment agreement or termination without cause, equal to 50% of his base salary at the time of termination, or (2) alternatively, in the event of a change in control of Ohr, upon (i) his termination without cause, (ii) expiration of the term of his employment agreement, or (iii) as a result of a constructive termination (that is, his resignation because he has reasonably determined in good faith that his titles, authorities, responsibilities, salary, bonus opportunities or benefits have been materially diminished, that a material adverse change in his working conditions has occurred, that his services are no longer required in light of Ohr’s business plan, or Ohr has breached his employment agreement) which occurs: (x) concurrently with the change in control, or (y) within 12 months of the change in control, he was entitled to receive (A) severance pay in an amount equal to $400,000, (B) the value of any accrued but unused vacation time, (C) the amount of all accrued but previously unpaid base salary through the date of termination, and (D) all of his then current employment benefits for the longer of twelve (12) months or the full un-expired term of his employment agreement. Mr. Backenroth had the right, for a period of 30 to 90 days following termination of his employment to exercise his Ohr options to the extent such options are otherwise vested and exercisable as of the date of termination. In connection with the Merger, Mr. Backenroth remained our Chief Financial Officer.
On May 22, 2019, Mr. Backenroth entered into a new employment offer letter to serve as our Chief Financial Officer effective upon the consummation of the Merger. Under the terms of the offer letter, which superseded Mr. Backenroth’s prior employment agreement with us, Mr. Backenroth will receive a base salary of $320,000 per annum, a signing bonus of $95,000, and a stock option to purchase 772,923 shares of our common stock, allocated from our incentive option pool at an exercise price of $5.39 per share (the “Backenroth Option”), and also be eligible to receive a discretionary annual bonus of up to 35% of his base salary. We paid Mr. Backenroth the signing bonus on July 26, 2019.
On May 15, 2020, the Company awarded a cash bonus to Mr. Backenroth in the amount of $132,000 for services rendered during the fiscal year ended September 30, 2019, which were not committed during such fiscal year and were approved by the Compensation Committee of the Board during May 2020.
Mr. Backenroth’s resignation was not a result of any disagreement with the Company or any matter relating to its accounting or financial policies or procedures. On September 30, 2021, the Company entered into a Separation Agreement with Mr. Backenroth, pursuant to which Mr. Backenroth is entitled to (i) continuation of Mr. Backenroth’s monthly base salary of $31,750.00 for a period of six months, (ii) immediate vesting of Mr. Backenroth’s stock option grant dated July 12, 2019 covering 772,923 shares of common stock, and (iii) an extension of Mr. Backenroth’s post-termination exercise period of his outstanding vested stock options, which totaled 790,423 shares, as of September 30, 2021to March 31, 2024.
Mr. Backenroth resigned from the Company on September 30, 2021.
William Mann, Ph.D.
On July 22, 2020, the Company entered into an offer letter with Dr. Mann. Pursuant to his offer letter, Dr. Mann’s annual salary will be $375,000, and he will have an annual performance bonus with a target of 40% of his base salary. Dr. Mann’s employment will be on an “at will” basis. Additionally, the Company granted Dr. Mann an option to purchase 175,000 shares of common stock under the Company’s 2019 Stock Incentive Plan. Subject to Dr. Mann’s continued employment with the Company, 1/4th of the shares underlying his option to purchase common stock will vest on the first anniversary of Dr. Mann’s start date, and 1/36th of the